The EU Medical Device Shake‑Up: What International Companies Should Prepare For
Overview
The EU medical devices regulatory framework continues to evolve, with significant legislative and policy reforms progressing in 2026. For companies bringing medical devices and in vitro diagnostics (IVDs) to the EU market, the landscape remains complex and resource intensive, but meaningful changes are now on the horizon.
Although the EU’s Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR), which came into force in 2021 and 2022 respectively, introduced stricter requirements compared to the previous Directives, such as on clinical evidence, post market surveillance, and economic operator requirements, their implementation has been challenging. The absence of grandfathering for legacy devices, limited notified body capacity, and strict rules without clear guidance have created backlogs, slowed innovation, and increased the risk of supply disruption. These pressures have prompted sustained criticism and repeated calls for reform.
For non-EU manufacturers, the stakes are even higher. Requirements include appointing an EU Authorized Representative, registering in EUDAMED, and ensuring compliance with EU-specific labelling and language requirements. Failure to plan early for notified body engagement and certificate renewals can result in missed market opportunities. The complexity and cost of compliance have led some companies to reconsider EU launches altogether, underscoring the importance of proactive strategy and resource allocation. Against this backdrop, proactive planning is increasingly critical to maintaining EU market access.
In March 2025, we published an Advisory on the latest developments and ongoing challenges with EU medical device legislation. Since then, the European Commission has released major new proposals to amend the MDR and IVDR, alongside reforms to notified body oversight and new transparency obligations tied to the phased rollout of EUDAMED. This Advisory summarizes the most important changes manufacturers should monitor and prepare for now.
Proposed Changes to MDR and IVDR
Towards the end of 2024, the European Commission launched a public consultation on the MDR and IVDR, framed as a “targeted evaluation” to identify problems with the legal framework. This was launched in part due to the European Parliament adopting a resolution on the urgent need to revise the MDR and the IVDR. Since then, on September 8, 2025, the European Commission published a call for evidence on “the targeted revision” of the regime, with the aim of identifying methods to tackle critical issues experienced throughout the industry caused by the regulations.
On December 16, 2025, the European Commission published its proposals on the amendment of the MDR and IVDR, marking a pivotal moment for the EU healthcare and MedTech landscape.
The proposals provide an overhaul of current requirements under the MDR and IVDR, aimed at streamlining regulatory processes, reducing administrative burden, and enhancing predictability, while maintaining patient safety and public health. For manufacturers, healthcare institutions, innovators, and industry stakeholders, the proposals signal a shift toward a more proportionate, risk-based system that supports timely access to critical technologies without compromising quality.
A summary of key proposals is set out below:
- Adaptive Pathways for Breakthrough and Orphan Devices: The proposals would create faster, priority assessment routes for qualifying “breakthrough” and “orphan” devices, via rolling reviews and expert panel input, enabling earlier EU market access. Notified bodies could issue certificates based on reduced pre market data and supported mandatory post market evidence generation. Expert panel involvement would be formalized, with a statutory basis for providing early scientific advice and supporting pre market evaluation. Together, these reforms aim to lower entry barriers for high innovation or high unmet need devices while maintaining a proportionate, risk based oversight approach.
- Streamlined Clinical Evidence and Equivalence: The proposals would reinstate a more flexible equivalence pathway for Class IIb and III devices, allowing manufacturers to rely on existing clinical data — including peer data and published literature — without requiring contracts with other manufacturers. This would reduce the need for new, resource intensive clinical investigations being conducted for well understood technologies and enable more proportionate evidence generation aligned with device risk.
- Extended Certificate Validity and Grandfathering: The proposals would allow certificates to remain valid indefinitely, with expiry applied only where a notified body identifies a justified risk based reason to limit validity. They would also enable certain legacy CE marked devices designated as orphan devices to remain on the EU market beyond the current transitional deadlines. These changes aim to reduce supply disruption, support continuity for established product portfolios, and provide non EU manufacturers with a more predictable and stable pathway for maintaining EU market access while planning longer term compliance strategies.
- Revised Classification Rules (Especially Software): At present, many software devices are classified within higher risk classes by default, often irrespective of their actual clinical impact. This has increased the need for notified body involvement and related costs. The European Commission proposes targeted revisions to the classification rules to better reflect actual risk, including reclassifying certain software and accessories into lower risk categories. Provided that a lower classification is possible (this may not be the case for the majority of software devices), manufacturers of software or accessories may be able to avoid costly assessments, reducing time and expenses.
- Improved Notified Body Governance: The proposals would introduce structured notified body-manufacturer dialogue, risk based assessment approaches, remote audit options, and a formal dispute resolution mechanism. These reforms aim to increase transparency, harmonize practices across Member States, and give manufacturers clearer and earlier insight into classification, evidence expectations, and conformity assessment pathways — ultimately improving predictability and reducing procedural delays.
- Regulatory Sandboxes and Digitalization: The proposals would establish EU level regulatory sandboxes to support evidence generation for innovative technologies, including AI driven and adaptive systems, by allowing controlled, real world, or simulated testing under regulatory oversight. They also introduce broader digitalization measures, such as enhanced digital declarations, clearer and more consistent cybersecurity requirements, and coordinated mechanisms for combined studies. These reforms aim to give innovators earlier clarity, more flexible evidence pathways, and a regulatory framework better suited to emerging technologies.
- International Reliance: The proposals introduce new provisions that would empower the EU to enhance regulatory cooperation with other authorities. By enabling the EU to rely on trusted third-country assessments (e.g., as part of the Medical Device Single Audit Program), companies may see reduced duplication of audits or certifications for manufacturers operating across multiple markets. While this does not change the requirement for full compliance with EU regulatory obligations, it may support greater international alignment over time.
- Reducing the Regulatory Burden on AI Medical Devices: The EU AI Act introduces a new regulatory framework for “high-risk AI systems,” which would apply to most AI medical devices. Manufacturers would need to comply with this framework in parallel with the MDR or IVDR requirements (as applicable). The proposals provide that AI medical devices, which are considered high-risk AI systems, would not need to comply with the AI Act in addition to the MDR or IVDR. AI-specific requirements may, however, be directly integrated into the EU medical device framework through Delegated or Implementing Acts. If adopted, it would represent a significant simplification of the rules for AI medical devices.
Next Steps
The proposals have been submitted to the European Parliament and Council for review. Once they have adopted their own positions on the text, negotiations to agree on a final text to be formally adopted will take place. At this stage, it is challenging to anticipate what the result of the political negotiations will be, and it is unclear when the new rules will take effect. However, please see below a potential timeline based on feedback collected from various stakeholders:
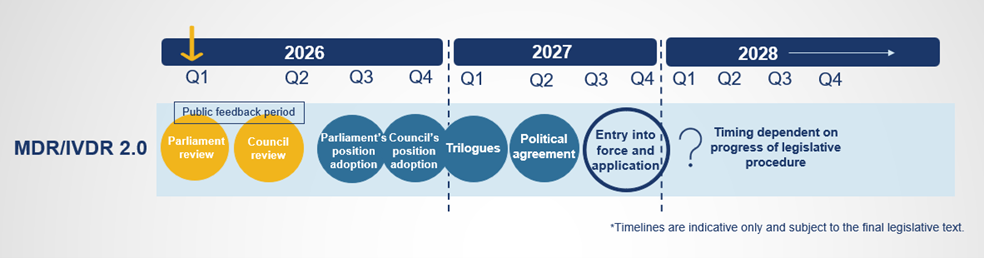
A feedback period has been opened, running from January 7, 2026 to March 5, 2026. All feedback received will be summarized by the European Commission and presented to the Parliament and Council with the aim of feeding into the legislative debate.
Note that the MDR and IVDR have not yet been amended; therefore, the transition periods under the MDR and IVDR, as described in our previous Advisory, should still be adhered to.
EU’s New Implementing Regulation for Notified Bodies
On December 12, 2025, the European Commission introduced a draft Implementing Regulation to standardize the quality management and procedural protocols for notified bodies under the MDR and IVDR.
The proposal directly addresses long standing pain points: opaque pricing, unpredictable timelines, and variable re certification practices across notified bodies. It would impose standardized quotation inputs (so you know exactly what information notified bodies need before they price), require detailed cost breakdowns (and justification for increases), set EU wide maximum timelines (e.g., 30 days for application review, 120 days for QMS audits, 90 days for product verification, and 15 days to issue certificates), and limit/structure “interruptions” during reviews to reduce serial back and forth. It also mandates notified body monitoring and public reporting on duration and costs, and codifies re certification content and clocks (60 days for notified body review; 15 days to issue renewed certificates). If adopted after consultation in 2026 as planned, these rules could materially improve predictability for manufacturers budgeting EU launches or MDR/IVDR certificate renewals. Please note that notified bodies are currently unhappy with the proposed timelines and are pushing to change them.)
What this means for you and steps you can take:
- Notified body selection becomes more strategic and more comparable. Use the forthcoming public performance and cost data to shortlist with the right designation scope and capacity for your technology class. Build these maximum timeline clocks into your internal EU launch or renewal plans and commercial commitments.
- Front load your submission package. Because the proposal caps and structures review interruptions, weak or incomplete files will be riskier; invest early in technical documentation, clinical/performance evidence, and QMS readiness to avoid burning your interruption “allowance.”
- Budget with fewer unknowns. Standardized quotations and cost variance justifications should reduce contingency buffers; still, retain some flexibility for expert panel/European Medicines Agency related pauses, which are handled separately under the draft proposal.
- Plan renewals earlier. With formalized re certification content and timelines, as well as a one year advance notification trigger, align your evaluation and safety reports, risk files, and state of the art updates well ahead of expiry to stay within the 60 and 15 day notified body clocks.
The measures would be phased in, respecting existing conformity agreements, with full annual reporting obligations commencing in 2028. The draft is currently open for public consultation until January 23, 2026, after which it will progress toward anticipated adoption in early 2026.
New Transparency Requirements
On November 27, 2025, the European Commission announced that four critical modules of EUDAMED — the EU’s centralized medical device and IVD database — are now fully functional, triggering a six-month transition period.
Starting May 28, 2026, manufacturers, importers, authorized representatives, notified bodies, and national authorities must comply with various transparency obligations under both the MDR and IVDR.
We have set out a compliance checklist for EU market entry in relation to these requirements:
1. Device Registration
- Register all new device models in EUDAMED before placing them on the EU market.
- Include unique device identifier/device identifier (UDI-DI), model details, and relevant conformity assessment information.
2. Economic Operator Registration
- Ensure your company, authorized representative, and importers are registered in EUDAMED.
- Obtain a Single Registration Number (SRN) for each economic operator.
3. Notified Body Certificates
- Upload new certificates issued by notified bodies into EUDAMED immediately.
- Plan for uploading historical certificates still valid by 2027 for full transparency.
4. Data Verification and Maintenance
- Implement internal processes for regular data verification in EUDAMED.
- Confirm that authorized representatives and importers validate your registration.
5. Transparency and Competitive Insight
- Prepare for public visibility of notified body certificates, which will be used for benchmarking.
- Consider monitoring competitor certifications for strategic positioning.
6. Operational Readiness
- Allocate resources for EUDAMED data entry and compliance monitoring.
- Train regulatory and quality teams on new EU requirements and timelines.
7. Timeline Awareness
- The transition period ends May 28, 2026; full compliance for new devices is required thereafter.
- Historical certificate uploads for devices where these are still placed on the market are due in 2027.
If you would like to discuss the current changes and how they may impact your product portfolio in the EU, please feel free to reach out to any of the authors or your existing Arnold & Porter contact. You can also follow developments on our BioSlice Blog.
© Arnold & Porter Kaye Scholer LLP 2026 All Rights Reserved. This Advisory is intended to be a general summary of the law and does not constitute legal advice. You should consult with counsel to determine applicable legal requirements in a specific fact situation.
Key Contacts



