Updated FDA Guidance Helps Device Study Sponsors Better Anticipate Coverage for Investigational Devices
On December 5, 2017, FDA published a final guidance document entitled "FDA Categorization of Investigational Device Exemption (IDE) Devices to Assist the Centers for Medicare and Medicaid Services (CMS) with Coverage Decisions" (Final Guidance).1 CMS reimbursement determinations for IDE products hinge on whether the device is deemed a Category A (experimental) or Category B (non-experimental/investigational) product. To this end, the Final Guidance further develops a longstanding dialogue between CMS and FDA, clarifying the criteria for initial categorization and the circumstances in which the categorization of an IDE product may change. Notably, the Final Guidance says that where a drug is added to a previously approved or cleared device, such a product typically would fall under Category A and thus would not receive coverage unless relevant new information is available to address initial questions of safety and effectiveness associated with addition of the drug. The Final Guidance also provides key examples of how data gathered through an Early Feasibility Study (EFS) or during a previous enrollment stage of an IDE that received a staged approval might be used to justify re-categorization of a device type from Category A to Category B. The Final Guidance provides important information that should be considered by medical technology companies developing cost recovery strategies for FDA-regulated medical device trials–particularly those with relatively long time-horizon studies.
Background
The Final Guidance builds on a regulatory framework that CMS and FDA have developed in collaboration. At a high level, devices that may be covered under Medicare include: (1) devices approved by FDA through the Pre-Market Approval (PMA) process; (2) devices cleared by FDA through the 510(k) process; (3) FDA approved IDE Category B devices; and (4) Hospital IRB-approved non-significant risk devices.2 For investigational devices that do not fall into Category B, Medicare does not reimburse for the investigational device but will pay for the routine costs associated with trials for the device.3 In September 1995, CMS (then known as the Health Care Financing Administration) published a final rule and entered into an Interagency Agreement (IA) with FDA that addressed reimbursement categorization of IDE devices.4 In the IA, CMS and FDA agreed on the criteria that FDA would use to categorize IDE devices. CMS would use these categorizations in turn to determine whether items and services were eligible for coverage under the "reasonable and necessary" threshold set by Social Security Act Section 1826(a)(1)(A).5
CMS modified the definitions of Category A and B devices through a December 10, 2013 Final Rule, published with FDA's concurrence, that centralized IDE review.6 Under the 2013 Final Rule, as codified at 42 C.F.R. 405.201(b):
- "Category A (Experimental) device refers to a device for which 'absolute risk' of the device type has not been established (that is, initial questions of safety and effectiveness have not been resolved) and the FDA is unsure whether the device type can be safe and effective."
- "Category B (Nonexperimental/investigational) device refers to a device for which the incremental risk is the primary risk in question (that is, initial questions of safety and effectiveness of that device type have been resolved), or it is known that the device type can be safe and effective because, for example, other manufacturers have obtained FDA premarket approval or clearance for that device type."
These definitions remain in place. In order to receive coverage under Medicare, an IDE study must meet certain criteria outlined in 42 C.F.R. 405.212.7 So long as CMS determines these criteria are met, if a device falls in Category A, Medicare will cover routine care items and services furnished in a FDA-approved Category A IDE study.8 For a Category B device, if CMS determines prior to the submission of the first related claim that the conditions of 42 C.F.R. 405.212 are met, Medicare will cover the IDE device itself as well as the routine care items and services furnished in an FDA-approved Category B IDE study.9
In the years following the 1995 IA, FDA expanded on this framework, issuing guidance to address categorization criteria relevant to Early Feasibility Studies (EFS) and executing a Memorandum of Understanding (MOU) with CMS to reflect both agencies' adoption of the changes under the 2013 Final Rule (the MOU supersedes the 1995 IA).
Content of the December 5, 2017 Final Guidance
The Final Guidance addresses existing gaps in this regulatory history by clarifying FDA's approach to the categorization criteria (including in the case of EFS) and by describing a pathway for changing categorization from Category A to Category B (or vice versa).
First, the Final Guidance walks through FDA's process for categorization. After receiving an IDE, FDA determines whether the sponsor has provided sufficient information to support initiation of a clinical study. If FDA determines there is adequate support for initiation of a human clinical study, no subject protection concerns preclude initiation of the investigation, and the benefit-risk profile is sufficiently favorable to justify enrollment, the IDE application is "approved" or "approved with conditions." To determine categorization (A or B), FDA uses a multi-step analysis captured in this flowchart (Appendix A of the Final Guidance):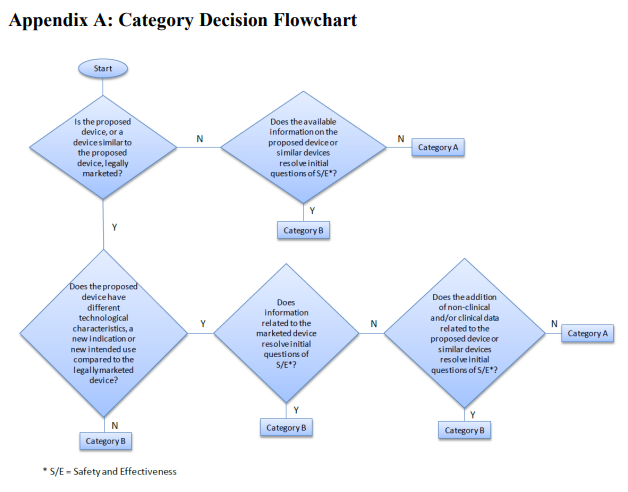
FDA explains it will assign a device to Category A if it meets one or more of the following three criteria:
- No PMA approval, 510(k) clearance, or De Novo request has been granted for the proposed device or similar devices, and data on the proposed device or other similar devices do not resolve initial questions of safety and effectiveness and FDA is unsure whether the device type can be safe and effective.
- The proposed device is being studied for a new indication, or new intended use, for which information from the proposed or a similar device related to the previous indication or intended use does not resolve initial questions of safety and effectiveness. Available non-clinical and/or clinical data on the proposed device or similar devices relative to the new indication or intended use also do not resolve these questions and FDA is unsure whether the device type can be safe and effective.
- The proposed device has different technological characteristics compared to a legally marketed device, and information related to the marketed device does not resolve initial questions of safety and effectiveness for the proposed device. Available non-clinical and/or clinical data on the proposed device or similar devices also do not resolve these questions and FDA is unsure whether the device type can be safe and effective.
FDA will assign a device to Category B, instead, if it meets one or more of the following three criteria:
- No PMA approval, 510(k) clearance, or De Novo request has been granted for the proposed device or similar devices; however, available information (e.g., feasibility study data) from the proposed device or a similar device resolve the initial questions of safety and effectiveness.
- The proposed device is being studied for a new indication or new intended use; however, information from the proposed or a similar device related to the previous indication or intended use resolves the initial questions of safety and effectiveness. In some cases, additional non-clinical and/or clinical data on the proposed device may also have been used to resolve these questions.
- The proposed device has similar technological characteristics compared to a legally marketed device, and information related to the marketed device resolves the initial questions of safety and effectiveness for the proposed device. In some cases, additional non-clinical and/or clinical data on the proposed device may also have been used to resolve these questions.
FDA emphasizes that a change in categorization can be initiated by FDA directly or in response to request from the sponsor, which may be submitted at any time as an IDE supplement (FDA will render a decision within 30 days). This flexibility accounts for the wide variability in sources and timing of data that might justify a change of categorization. The guidance describes several circumstances under which a device might be reclassified from Category A to Category B:
- Where a completed study in which the device was designated as Category A resulted in clinical data that resolved initial questions of safety and effectiveness;
- When an IDE receives a staged approval or staged approval with conditions, and where data gathered from the first or subsequent enrollment stages resolves initial questions of safety and effectiveness for the device category;
- When a sponsor submits an IDE supplement that includes data supporting a categorization change; this data may include but is not limited to: peer-reviewed studies on the same or a similar device, premarket or postmarket data from ex-US studies on the same or a similar device, reference to commercialization of a device of similar type, preliminary clinical data on the device (e.g., initial data from a staged study or feasibility study), or additional non-clinical data on the same or a similar device.
FDA notes that in order for a change to be made in the reverse direction, from Category B to Category A, "there should be directly relevant new information which demonstrates that initial questions of safety and effectiveness have not been resolved" (for example, a comparison of the proposed device to a commercialized device of a similar type, accompanied by an explanation of why initial questions of safety and effectiveness for the proposed device remain that justify the categorization).
Examples of Categorization Under the Final Guidance
In pages 11-13 of the Final Guidance, FDA provides several examples of when a Category A or B determination would be appropriate, or where a change from A to B would be warranted. Most notably, FDA explains that a Category A determination is warranted where "[a] drug is added to a previously approved or cleared device. While substantial information is known about the previously approved or cleared device, the drug has not previously been used on this type of device, and relevant non-clinical or clinical data are not available to address the initial questions of safety and effectiveness associated with the addition of the drug." 10
FDA provides several additional (non-exclusive) examples of cases where a Category A determination may be appropriate. For example:
- Where a device is completely novel and has no, or limited, previous human use and there are initial questions of safety and effectiveness. There is adequate non-clinical information to support initiation of an early feasibility study that will provide data to inform potential device design or procedural improvements.
- Where an already-approved or already-cleared device is being evaluated for a new intended use or indication for which the device will be placed in a different anatomical location. The device's technology is unchanged from what was initially approved or cleared. While some non-clinical data on this device may be used to anticipate certain aspects of device performance, it is still uncertain as to whether the device can be safely placed, and be effective, in the new anatomical location. Therefore, there are inadequate data to resolve the initial questions of safety and effectiveness relative to the new intended use or indication, and FDA is unsure whether or not the device type can be safe and effective.
In addition to the above, FDA provides examples in which initial questions of safety have been answered through available data, but not questions of effectiveness —or vice versa.11
Similarly, the Final Guidance walks through examples of where a Category B determination would be warranted:
- The insertion system of an approved device has been modified to improve ease of use for the clinician. Non-clinical test data resolved initial questions of safety and effectiveness related to this change; however, confirmatory clinical information about the device performance is required due to the inherent differences between the non-clinical test environment and the clinical setting.
- A new device will be studied for an indication for which substantial safety and effectiveness information exists from other similar device(s) of the same type that are used for the same or a similar indication. Clinical information from similar devices and non-clinical test data for the new device that have been provided can answer initial safety and effectiveness questions regarding this indication.
- New device sizes will be added to a product matrix for an approved device. Initial questions of safety and effectiveness have been resolved based on experience with the approved device, and it is generally understood how the new device sizes will perform. The new device sizes will be evaluated in a clinical study such that confirmatory safety and effectiveness information relevant to these sizes can be gathered.
Other key examples include where adequate data from non-clinical testing and the clinical results of a feasibility study resolve initial questions of safety and effectiveness; or where an approved device is evaluated in a new patient population or for a new indication and relevant data exists to resolve initial questions of safety and effectiveness.12
FDA also provides the following examples of when a device would be moved from Category A to B (FDA notes this is not an exclusive list):
- A novel insertion procedure will be used to place an already-approved or -cleared device, and there are initial questions of safety and effectiveness regarding the novel insertion procedure that have not been resolved. In this case, these questions of safety and effectiveness may be answered in a short timeframe with a limited number of subjects in the context of a larger clinical study. Therefore, the device will be evaluated in a staged clinical study where the first stage falls under Category A, but the device may be re-categorized to Category B if initial questions of safety and effectiveness are resolved as the study continues.
- Adequate data have been gathered on a device from non-clinical testing and an early feasibility study has been conducted within the U.S., such that initial questions of safety and effectiveness have been resolved. Additional data are needed to help inform a pivotal study design; therefore, a traditional feasibility study will be initiated. The data gathered in EFS justify the change from category A to B.
- A device is currently being evaluated in a clinical study and has been designated Category A. While the study is being conducted, clinical study results for similar devices become available which resolve initial questions of safety and effectiveness for the device.
Compliance Ramifications of Classification and Coverage Determinations
Medicare coverage can have significant business and compliance ramifications for a sponsor company. Medicare coverage creates an opportunity for cost recovery for research that might otherwise be cost-prohibitive for a sponsor to initiate and allows companies to generate income that can be used to invest in technology improvements and to build commercial and clinical support in anticipation of commercial launch. While cost recovery through reimbursement creates potential business opportunities, it also means that clinical trial-related costs–such as payments to physician-investigators and their institutions–are more likely to come under scrutiny under the Anti-Kickback Statute, False Claims Act, and related laws. For example, the provision of a free or heavily discounted test device in a Category B trial raises potential fraud and abuse concerns, as it can create a perception that the sponsor is creating inappropriate financial incentives for the physician-investigator and their institution to select the use of the investigational device. Therefore, where Medicare payment is available following an FDA categorization, sponsors should ensure that they have an understanding of the relevant Anti-Kickback Statute Safe Harbors relevant to the specific contracting arrangements in their trials (e.g., safe harbors for product discounts, personal services, and equipment rentals). Medicare coverage also triggers federal transparency reporting obligations for sponsors under the Physician Payments Sunshine Act.13 Cost-sharing payments between a sponsor and a clinical trial subject can also raise Medicare Secondary Payor reporting and compliance concerns under Section 111 of the Medicare, Medicaid, and SCHIP Extension Act of 2007 (MMSEA) as well as triggering provider compliance requirements (such as a need to send advance beneficiary notices to trial subjects). Where test article costs will not be covered by CMS, sponsors must consider whether charging investigators or institutions is in line with FDA regulations and policy for charging for investigational devices.14 The availability of reimbursement does not obviate the need to consider prohibitions on pre-approval marketing and both FDA and HHS-OIG have raised concerns with the use of research arrangements as a means of market "seeding" or as a means of rewarding past, current, or future referral decisions. Finally, sponsors must consider how the availability of Medicare reimbursement will affect cost recovery from potential research subjects, and whether informed consent forms adequately set forth the financial obligations subjects will bear, including for any injuries resulting from a Category A or B study.
Conclusion
This Final Guidance provides additional clarity as to how FDA will handle categorization and re-categorization of devices from Category A to B and updates prior FDA policy in this area. The guidance is particularly informative in the case of IDE devices subject to EFS, or in cases where the generation of additional data (for example in a staged clinical study) resolves initial questions of safety and effectiveness, justifying a change from Category A to Category B. The guidance also offers practical examples to sponsors and is a reminder that availability of reimbursement is an essential part to cost recovery and budget planning for a prospective clinical trial. Finally, though not addressed by this guidance, sponsors must keep in mind that the business advantages available through favorable FDA categorization determinations must be balanced with the legal and compliance ramifications of the resulting CMS determination, including with regard to concepts such as fair market value for investigator payments, cost-sharing considerations for Medicare beneficiaries, and transparency reporting requirements.
© 2017 Arnold & Porter Kaye Scholer LLP. This Advisory is intended to be a general summary of the law and does not constitute legal advice. You should consult with counsel to determine applicable legal requirements in a specific fact situation.
-
See Guidance for Sponsors, Clinical Investigators, Industry, Institutional Review Boards, and Food and Drug Administration Staff.
-
CMS, Medicare Benefit Policy Manual § 10 (Rev. 198, 11-06-14).
-
CMS, Medicare National Coverage Determinations (NCD) Manual § 310.1 (Rev. 198, 06-29-17).
-
See 60 Fed. Reg. 48417 (Sept. 19, 1995) (including, as appendix, the IA).
-
Under the IA, Category A devices were those for which no marketing application had been approved through the pre-marketing approval (PMA) process for any indication or devices that would otherwise be in Category B but had undergone significant modification for a new indication or use. Category B devices were those under investigation to demonstrate substantial equivalence to a legally marketed device through the 510(k) process or devices comparable to a MPA-approved device.
-
78 Fed. Reg. 74230 (Dec. 10, 2013).
-
These include: "(1) The principal purpose of the study is to test whether the device improves health outcomes of appropriately selected patients. (2) The rationale for the study is well supported by available scientific and medical information, or it is intended to clarify or establish the health outcomes of interventions already in common clinical use. (3) The study results are not anticipated to unjustifiably duplicate existing knowledge. (4) The study design is methodologically appropriate and the anticipated number of enrolled subjects is adequate to confidently answer the research question(s) being asked in the study. (5) The study is sponsored by an organization or individual capable of successfully completing the study. (6) The study is in compliance with all applicable Federal regulations concerning the protection of human subjects found at 21 CFR parts 50, 56, and 812 and 45 CFR part 46. (7) Where appropriate, the study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Studies of all medical technologies measuring therapeutic outcomes as one of the objectives may be exempt from this criterion only if the disease or condition being studied is life threatening and the patient has no other viable treatment options. (8) The study is registered with the National Institutes of Health's National Library of Medicine's ClinicalTrials.gov. (9) The study protocol describes the method and timing of release of results on all pre-specified outcomes, including release of negative outcomes and that the release should be hastened if the study is terminated early. (10) The study protocol must describe how Medicare beneficiaries may be affected by the device under investigation, and how the study results are or are not expected to be generalizable to the Medicare beneficiary population. Generalizability to populations eligible for Medicare due to age, disability, or other eligibility status must be explicitly described." 42 C.F.R. 405.212(a).
-
-
-
Final Guidance at 11 (emphasis added).
-
See page 11-12 of the Final Guidance.
-
For the full list, see page 12 of the Final Guidance.
-
Section 6002 of the Affordable Care Act, 42 U.S.C. § 1320a-7a.
-
The Investigational Device Exemption (IDE) regulations allow sponsors to charge for an investigational device, however, the charge should not exceed an amount necessary to recover the costs of manufacture, research, development, and handling of the investigational device (21 CFR 812.7(b)). A sponsor justifies the proposed charges for the device in the IDE application, states the amount to be charged, and explains why the charge does not constitute commercialization (21 CFR 812.20(b)(8)). FDA generally allows sponsors to charge investigators for investigational devices, and this cost usually is passed on to the subjects.
Key Contacts
