FDA, State, and Local Governments Act to Modernize the Regulation of Sunscreen Products in the United States
On February 26, 2019, FDA published its long-awaited proposed rule, Sunscreen Products for Over-the-Counter Human Use (Proposed Rule).1 The Proposed Rule is a step towards FDA's full compliance with the mandates of the 2014 Sunscreen Innovation Act (SIA)2 and builds on the stayed 1999 Sunscreen Final Monograph3 by proposing to, among other things, formally classify the safety and effectiveness of certain active ingredients and dosage forms, update sunscreen testing and recordkeeping requirements, and address new uses of sunscreens, including the sale of combination sunscreen-insect repellant products. FDA has proposed a 90-day comment period, and comments must be submitted on or before May 28, 2019.
In addition to updating testing and recordkeeping requirements, FDA proposes to:
- classify zinc oxide and titanium dioxide as Category I (Generally Recognized as Safe and Effective – GRASE) sunscreen active ingredients, including certain products made with nanotechnology;
- designate oil, lotion, cream, gel, butter, paste, ointment, stick, and spray sunscreen dosage forms as GRASE under the monograph (and the powder form potentially GRASE subject to FDA's receipt of additional safety and efficacy data regarding the dosage form);
- designate all other dosage forms, including wipes, towelettes, body washes, and shampoos as "new drugs" that are ineligible for inclusion in the monograph;
- establish new labeling and broad spectrum requirements (including a maximum SPF level of SPF 60+); and
- classify combination insect repellant-sunscreen products as Category II (not GRASE).
Below, we provide an overview of the key provisions included in the Proposed Rule as well as other relevant state and city-level measures banning certain sunscreen active ingredients in Hawaii and Key West, FL. Given the increase in use of sunscreen ingredients in topical consumer products, including moisturizers and concealers, these developments are sure to impact any stakeholder in the OTC drug or personal care product industry.
I. Background
The Proposed Rule is FDA's most recent effort to modernize the regulation of sunscreen products in the US to account for increasing use of sunscreen in consumer products. The Proposed Rule builds on numerous Federal Register notices issued over the past decade4 and attempts to close the loop on the stayed 1999 Sunscreen Final Monograph by meeting the SIA's requirement to issue a final OTC sunscreen monograph by November 29, 2019. The SIA was passed in 2014 and amended the Federal Food, Drug, and Cosmetic Act (FD&C Act) to expedite the review of nonprescription sunscreen active ingredients that were not included in the stayed 1999 Sunscreen Final Monograph. While the SIA required issuance of this Proposed Rule (plus certain guidance documents), the sunscreen active ingredients submitted under the procedures established in FDA's Time and Extent Application (TEA) notice that are being considered under processes established by the SIA are not under consideration in the Proposed Rule.5
II. Overview of Proposed Sunscreen Rule
There are seven key takeaways in the Proposed Rule for stakeholders—(1) how FDA intends to classify 16 sunscreen active ingredients; (2) FDA's position on specific sunscreen dosage forms; (3) FDA's perspective on the use of nanomaterials and nanotechnology in sunscreen products; (4) FDA's proposed maximum labeled SPF values and broad spectrum requirements; (5) recommended labeling changes, including revisions to the products' statement of identity; (6) proposed modifications to sunscreen testing and recordkeeping requirements; and (7) FDA's proposal to classify combination insect-repellant sunscreen products as not GRASE.
A. Classification of Active Ingredients
Central to the Proposed Rule is FDA's safety assessment of sixteen sunscreen active ingredients that were originally included in the stayed 1999 Sunscreen Final Monograph. Following a review of publicly available data, including, but not limited to scientific literature, adverse event reports, submissions to the sunscreen monograph docket, FDA Advisory Committee Reports, and FDA and European Medicines Agency approval documents, FDA is proposing to classify the active ingredients as follows:
| Category I (GRASE) | Zinc Oxide, Titanium Dioxide |
| Category II (Not GRASE) | Para-Aminobenzoic Acid (PABA), Trolamine Salicylate |
| Category III (Insufficient Data Available to Permit Final Classification) | Avobenzone, Cinoxate, Dioxybenzone, Ensulizole, Homosalate, Meradimate, Octinoxate, Octisalate, Octocrylene, Oxybenzone, Padimate O, Sulisobenzone |
Category I – GRASE
FDA proposes to classify zinc oxide and titanium dioxide as GRASE. For both active ingredients, FDA's determination is based, in no small part, on existing and substantial evidence that these mineral compounds largely do not penetrate the skin (even particles on the nanoscale (1-100 nanometers)), and to the extent that "de minimis" penetration occurs, do not cause adverse health effects.6
Category II – Not GRASE
FDA proposes to classify para-aminobenzoic acid (PABA) and trolamine salicylate as not GRASE because the Agency has determined that the risks outweigh the benefits for the use of these active ingredients in sunscreen products. This determination should not affect the marketplace for sunscreen products or manufacturers in any measureable way because to FDA's knowledge, neither active ingredient is currently being marketed in sunscreen products in the United States.
Category III – Insufficient Data Available to Permit Final Classification
FDA proposes to classify cinoxate, dioxybenzone, ensulizole, homosalate, meradimate, octinoxate, octisalate, octocrylene, padimate O, sulisobenzone, oxybenzone, and avobenzone as active ingredients for which the public record lacks sufficient information to support a positive GRASE determination at this time. According to the Proposed Rule, "significant" data gaps exist for many of these active ingredients. For the active ingredients with significant data gaps (all except oxybenzone and avobenzone), FDA expects that data from clinical studies (e.g., human dermal safety studies and human absorption studies/maximal usage trial (MUsT)), nonclinical studies, and postmarketing safety information (including the evaluation of adverse events reported during the commercial marketing of sunscreens) will be necessary to support a positive GRASE finding.7 FDA expects that supporting a positive GRASE finding for oxybenzone and avobenzone will require fewer studies and data, and has set forth specific data requests regarding these two ingredients (e.g., providing carcinogenicity data and conducting MUsT studies).8
Parties interested in submitting information regarding proposed Category III active ingredients will need to work quickly—some of the recommended studies take anywhere from six months to two years to complete. Should manufacturers need additional time to provide safety or effectiveness data for a particular sunscreen active ingredient, FDA has expressed willingness to "consider requests to defer further rulemaking with respect to a specific sunscreen active ingredient to allow for the submission of new safety and/or effectiveness data to the record if such requests are submitted to the docket within the initial 90-day comment period."9
B. Sunscreen Dosage Forms
In 2011, FDA issued an Advance Notice of Proposed Rulemaking, requesting data and information about sunscreen dosage forms.10 In this document, FDA outlined the dosage forms that it believed were eligible for inclusion in a sunscreen monograph based on their history of marketing before the OTC Drug Review started in 1972, and dosage forms that would not be eligible for inclusion in such a monograph. FDA also solicited information about the safety and effectiveness of spray dosage forms. Following a review of the comments to the docket, plus additional information on the marketing history of sunscreen powder dosage forms, FDA has proposed to classify the following sunscreen dosage forms as Category I (GRASE): oil, lotion, cream, gel, butter, paste, ointment, stick, and spray.
FDA has requested additional safety and efficacy data on the powder dosage form, though it is eligible for inclusion in the OTC sunscreen monograph. By contrast, FDA plans to consider all other dosage forms, including wipes, towelettes, body washes, and shampoos to be "new drugs" that are ineligible for inclusion in the OTC sunscreen monograph. FDA has taken the position that these dosage forms cannot be included in the monograph because, according to the proposed rule, FDA has no evidence that they were marketed prior to 1972.
While FDA has proposed that spray dosage forms are GRASE, due to the potential respiratory health risks from inhaling spray sunscreen and the potential flammability risk,11 FDA intends to impose formulation (particle size) limitations, labeling requirements, and particle size testing requirements on spray dosage forms in the final monograph. Regarding particle size, FDA proposes that 90% of the particles dispensed from a spray sunscreen must be at least 10 μm, while the other 10% must be 5 μm or greater.12 These same particle size limitations will also apply to powder dosage forms due to the risk of respiratory harm from inhalation. FDA also addressed concerns about the inhalation risk posed by the use of nanomaterials in spray sunscreens, but declined to propose conditions of use on nanomaterials in spray sunscreens because nanomaterials would not pass the 5 μm particle size requirement. The required particle size testing would be incorporated into the lot release testing that manufacturers perform as part of their current good manufacturing (cGMP) responsibilities under 21 CFR part 211.
C. Nanomaterials & Nanotechnology
In the Proposed Rule, FDA touches on the issue of sunscreens manufactured using nanotechnology or incorporating nanomaterials, particularly in reference to the proposed GRASE active ingredients and particle size requirements for spray sunscreens. As conveyed in the Proposed Rule, the Agency is not proposing to classify sunscreen products as Category I (GRASE) or Category II (not GRASE) based on the use of nanotechnology or nanomaterials. Nor is FDA planning to establish regulatory definitions of nanotechnology, nanomaterials, or nanoscale in this context. It does, however, intend to apply the considerations described in FDA's June 2014 guidance for industry, Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology, to sunscreen products.13 Additionally, FDA has specifically solicited comments about the use of nanomaterials and sunscreens, including: "specific nanomaterials or types of nanomaterials that have been used or proposed for use in OTC sunscreen products; concerns about sunscreen product safety, effectiveness, or quality associated with the use of nanomaterials in OTC sunscreen products, with supporting data;" the need for certain specifications or limitations for the use of nanomaterials in OTC sunscreen; particular nanomaterials that should not be used in OTC sunscreen, plus supporting scientific data, and comments on FDA's posture towards the use of nanomaterials in OTC sunscreen.14
D. Proposed Maximum SPF, Labeling Ranges and Broad Spectrum Requirements
SPF Maximum & Labeling Ranges
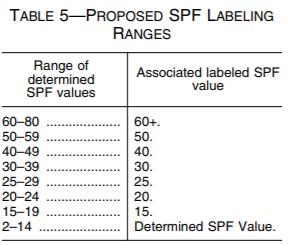
FDA is proposing to raise the maximum labeled SPF value for sunscreen products to SPF 60+. While FDA is proposing to permit sunscreen formulation up to SPF 80, manufacturers will not be able to convey that their products provide more SPF protection than 60+. The purpose of allowing a greater formulation value than labeled value is to account for the variability in SPF test results and to facilitate the development of products with improved UVA protection.15 Additionally, FDA proposes to require that sunscreens' labeled SPF values be based on a range of determined SPF values, as depicted in Table 5 below.
Broad Spectrum Requirement
FDA proposes to require that all sunscreens with a labeled SPF value of 15 and above provide broad spectrum protection, meaning protection from both UVA and UVB radiation. Sunscreens with a labeled SPF value below 15 need not provide broad spectrum protection. In fact, FDA is requesting comments on whether sunscreens with labeled SPF values from 2-14 should continue to be marketed. To qualify as broad spectrum, the sunscreen must "pass" the broad spectrum test, meaning that the product has a mean critical wavelength of 370nm or greater (a current requirement) and has a mean UVA I/UV ratio of 0.7 or higher (proposed requirement). This new test criteria should help ensure that sunscreens "have a more uniform amount of radiation protection across the UVA I, UVA II, and UVB ranges" and "eliminate the current potential for a product labeled as broad spectrum that has a higher SPF value to provide (unbeknownst to the consumer) poorer broad spectrum protection than a product labeled as broad spectrum with a lower SPF value."16
E. Labeling – Statement of Identity
Although FDA regulations currently require a statement of identity,17 the Proposed Rule sets forth specific requirements for sunscreen and sunscreen/skin protectant drug products. Per the technical requirements set forth in the Proposed Rule (including font size and placement requirements), the principal display panel (PDP) must display the statement of identity (SOI) as follows: the name of each sunscreen active ingredient listed in alphabetical order, followed by "Sunscreen" and the dosage form.18
F. Testing & Recordkeeping
FDA's regulations currently require final formulation testing to substantiate the labeled SPF values, water resistance statements, and broad spectrum statements.19 Citing concerns about current practices employed in final formulation testing, the Proposed Rule modifies the regulations governing testing to clarify the applicable requirements, improve study reliability, and to add protections for human subjects involved in the testing. Notably, the Proposed Rule clarifies that FDA's regulations pertaining to informed consent (21 CFR part 50) and IRB approval of research (21 CFR part 56) apply to clinical final formulation testing set forth in proposed 21 CFR 201.327(i).20 Additionally, the Proposed Rule imposes new recordkeeping requirements for final formulation testing (which FDA understands to be a manufacturing activity subject to cGMP requirements) that are aligned with requirements for other types of manufacturing subject to cGMPs.21 The Proposed Rule outlines who should retain records, the types of records that must be kept, and FDA's right to access testing records.22 It is particularly important for regulated industry to pay attention to these requirements because the Proposed Rule imposes penalties of misbranding for failure to comply with any of the provisions in proposed 21 CFR 201.327 (including failure to conduct final formulation testing in a registered establishment), and FDA may find that a product is adulterated for failure to comply with cGMPs or recordkeeping requirements in proposed 21 CFR 201.327.23
To clarify the responsibility for complying with the final formulation testing and recordkeeping requirements, the Proposed Rule creates a new term – "Responsible Person" – defined as the "manufacturer, packer, or distributor whose name appears on the labeling of a sunscreen product covered by this section."24 The Responsible Person's obligations are modeled after the regulatory obligations applicable to an investigational new drug (IND) application sponsor, and incorporate certain investigator responsibilities, yet are tailored to the specific sunscreen clinical and nonclinical formulation testing. For example, the Responsible Person must "select appropriately qualified personnel to conduct testing, ensure compliance with the requirements for IRB review and obtaining informed consent, and monitor the compliance of personnel with investigators' statements[,]" as well as comply with the new recordkeeping requirements.25 A Responsible Person may transfer all of its responsibilities under part 327 to another entity (e.g., a contract research organization and/or testing laboratory), except for recordkeeping requirements.
G. Combination Insect-Repellent and Sunscreen Products
Products containing sunscreens and insect repellants are subject to regulation by FDA under the FD&C Act, and the Environmental Protection Agency as pesticides under the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA). FDA has a number of concerns about these combination products, including how the interactions between sunscreen active ingredients and insect repellants may affect the safety and efficacy of the combined product. Following a review of the scientific literature, publicly-available EPA regulatory documents, and comments submitted in response to a "2007 Call for Data", FDA proposes to classify these products as Category II (not GRASE), and misbranded because "conflicting labeling requirements for their sunscreen and insect repellent components cannot be reconciled to create labeling that will sufficiently ensure safe and effective use of the sunscreen, as well as adequate directions for use as a sunscreen . . ."26
III. Request for Comments & Compliance Deadline
As noted, FDA has proposed a 90-day comment period, and comments must be submitted on or before May 28, 2019. FDA may consider requests to defer further rulemaking for a specific sunscreen active ingredient to allow for the submission of additional safety and/or efficacy data if such requests or comments are received within this 90-day comment period. If a sunscreen active ingredient is deferred, FDA expects that products containing that active ingredient can stay on the market while data is being gathered and studies are ongoing.27 Commissioner Gottlieb recently tweeted that the Agency "plan[s] to finalize the sunscreen proposed rule for the non-deferred ingredients and the other aspects of the rule. Once we receive data on a deferred ingredient and have a chance to consider such data, we intend to issue a final rule with our determination for that ingredient."28
FDA anticipates requiring full compliance with the final regulations one year after the effective date of the final rule, which could be as early as November 26, 2020. Note that the Agency does not intend to require full compliance with the final regulations for products introduced or delivered into interstate commerce prior to the compliance date (e.g., products in retail stores).
IV. State and City-Level Measures Banning Sunscreen Active Ingredients
Aside from measures at the federal level to address sunscreen active ingredient safety for use in humans, states are implementing (or considering) measures to prohibit certain sunscreen active ingredients due to the risk of environmental harm. For example, Hawaii's legislature determined that oxybenzone and octinoxate were significantly harmful to Hawaii's marine environment and ecosystems, and were damaging coral reefs and pursued legislation to mitigate this problem.29 Subsequently, on July 3, 2018, Hawaii's governor signed SB 2571, Act 104 into law, prohibiting the sale, offer for sale, or distribution for sale in Hawaii of any SPF sunscreen products contain oxybenzone and octinoxate starting on January 1, 2021.30 California's legislature attempted to follow in Hawaii's footsteps, introducing Assembly Bill 60, a bill to prohibit the sale of sunscreens containing octinoxate or oxybenzone without a prescription.31 Likewise, the city of Key West, Florida recently voted to ban the sale of sunscreens containing oxybenzone and octinoxate starting in January 2021, citing harm to coral reefs.32
* * *
Our team has carefully reviewed the Proposed Rule and the related state and local developments impacting the sale of products that contain sunscreen ingredients. We will continue to monitor developments in this area. In the interim, please feel free to contact us with any questions about the topics discussed in this advisory.
© Arnold & Porter Kaye Scholer LLP 2019 All Rights Reserved. This Advisory is intended to be a general summary of the law and does not constitute legal advice. You should consult with counsel to determine applicable legal requirements in a specific fact situation.
-
Sunscreen Products for Over-the-Counter Human Use, 84 Fed. Reg. 6204, Feb. 26, 2019.
-
21 USC § 360fff-5. The SIA requires FDA to issue a final OTC sunscreen monograph within five years of enactment of the SIA (by Nov. 29, 2019).
-
Sunscreen Drug Products For Over-The-Counter Human Use; Final Monograph, 98 Fed. Reg. 27666, May 21, 1999 (stayed on Dec. 31, 2001, 66 Fed. Reg. 67485).
-
Sunscreen Drug Product for Over-the-Counter Human Use; Request for Data and Information Regarding Dosage Forms, 76 Fed. Reg. 35669 (June 17, 2011).
-
The status and next steps for these active ingredients is available here. Note that the Proposed Rule does not address the safety of or establish a pathway for considering sunscreen active ingredients that are approved in other countries.
-
Note, FDA is not planning to impose any conditions on the use of nanomaterial forms of these active ingredients. 84 Fed. Reg. at 6216.
-
FDA noted that data from MUsT studies "should be properly validated according to good laboratory practices (21 CFR part 58). 84 Fed. Reg. at 6214.
-
-
-
Sunscreen Drug Product for Over-the-Counter Human Use; Request for Data and Information Regarding Dosage Forms, 76 Fed. Reg. 35669 (June 17, 2011).
-
The risk is posed by, for example, exposure to an open flame before the spray sunscreen has dried.
-
Based on the particle size data submitted to the 2011 ANPR docket, FDA believes that this limitation would not "unduly burden" sunscreen manufacturers.
-
84 Fed. Reg. at 6215, citing FDA Guidance for Industry, Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology, June 2014. Specifically, FDA intends to apply these two points from the guidance in particular: "(1) Whether a material or end product is engineered to have at least one external dimension, or an internal or surface structure, in the nanoscale range (approximately 1 nm to 100 nm); (2) Whether a material or end-product is engineered to exhibit properties or phenomena, including physical or chemical properties or biological effects, that are attributable to its dimension(s), even if these dimensions fall outside the nanoscale range, up to 1 micrometer (mm) (1,000 nm)."
-
-
-
-
-
-
-
-
-
84 Fed. Reg. at 6242-6243, 6271.
-
84 Fed. Reg. at 6242, 6264, 6271.
-
84 Fed. Reg. at 6241. The purpose of creating this definition is to help FDA assess compliance with section 327 because "it creates a chain of responsibility that is immediately apparent from the product's labeling.
-
-
-
Tweets of Scott Gottlieb, M.D., Mar. 2, 2019, 10:54 a.m.
-
Tweets of Scott Gottlieb, M.D., Mar. 2, 2019, 10:55 a.m.
-
Hawaii SB 2571, Act 104.
-
Press Release, Governor David Ige Signs Bill Making Hawaii First in the World to Ban Certain Sunscreens, Jul. 3, 2018; Hawaii SB 2571, Act 104.
-
California Legislature, Assembly Bill No. 60. Assembly Bill 60 was recently amended into an unrelated bill; thus, California likely will not seek to address this issue on the statewide level this year. Further action on this legislation may be pursued in 2020, depending to some extent on FDA's determination of active ingredient safety.
-
Washington Post, "We Have One Reef": Key West Bans Popular Sunscreens to Help Keep Coral Alive, Feb. 6, 2019.
Key Contacts

